
Reglas de prioridad de Cahn – Ingold – Prelog - Cahn–Ingold–Prelog priority rules

Las reglas de secuencia de Cahn-Ingold-Prelog ( CIP ) , llamadas así en honor a los químicos orgánicos Robert Sidney Cahn , Christopher Kelk Ingold y Vladimir Prelog , también llamadas reglas , sistema o convenciones de prioridad CIP , son un proceso estándar utilizado en química orgánica para y nombrar inequívocamente un estereoisómero de una molécula. El propósito del sistema CIP es asignar un descriptor R o S a cada estereocentro y un descriptor E o Z a cada doble enlace para que la configuración de la molécula completa se pueda especificar de forma única al incluir los descriptores en su nombre sistemático. Una molécula puede contener cualquier número de estereocentros y cualquier número de dobles enlaces , y cada uno normalmente da lugar a dos posibles isómeros. Una molécula con un número entero n que describe el número de sus centros estereogénicos normalmente tendrá 2 n estereoisómeros y 2 n -1 diastereómeros, cada uno de los cuales tendrá un par asociado de enantiómeros. Las reglas de secuencia CIP contribuyen a la denominación precisa de cada estereoisómero de cada molécula orgánica y organometálica con todos los átomos de ligadura de menos de 4 (pero incluida la ligadura de 6 también, este término se refiere al "número de átomos vecinos" unidos a un centrar).
El artículo clave que establece las reglas de secuencia del CIP se publicó en 1966, y fue seguido por más refinamientos, antes de incorporarse a las reglas de la Unión Internacional de Química Pura y Aplicada (IUPAC), el organismo oficial que define la nomenclatura orgánica , en 1974. Desde entonces, las reglas se han revisado, la última vez en 2013, como parte del libro de la IUPAC Nomenclatura de química orgánica . La presentación de la IUPAC de las reglas constituye el estándar oficial y formal para su uso, y señala que "el método ha sido desarrollado para cubrir todos los compuestos con ligadura hasta 4 ... y ... [extendido al caso de] ligadura 6 ... [así como] para todas las configuraciones y conformaciones de dichos compuestos ". Sin embargo, aunque la documentación de la IUPAC presenta una introducción completa, incluye la advertencia de que "es esencial estudiar los artículos originales, especialmente el artículo de 1966, antes de usar la regla de secuencia para casos que no sean bastante simples".
Un artículo reciente aboga por cambios en algunas de las reglas (reglas de secuencia 1b y 2) para abordar ciertas moléculas para las que los descriptores correctos no estaban claros. Sin embargo, sigue existiendo un problema diferente: en casos raros, dos estereoisómeros diferentes de la misma molécula pueden tener los mismos descriptores de CIP, por lo que es posible que el sistema CIP no pueda nombrar un estereoisómero sin ambigüedades, y pueden ser preferibles otros sistemas.
Pasos para nombrar
Los pasos para nombrar moléculas usando el sistema CIP a menudo se presentan como:
- Identificación de estereocentros y dobles enlaces ;
- Asignación de prioridades a los grupos adjuntos a cada estereocentro o átomo de doble enlace; y
- Asignación de descriptores R / S y E / Z.
Asignación de prioridades
Los descriptores R / S y E / Z se asignan utilizando un sistema para clasificar la prioridad de los grupos adjuntos a cada estereocentro. Este procedimiento, a menudo conocido como reglas de secuencia , es el corazón del sistema CIP. La descripción general de esta sección omite algunas reglas que solo son necesarias en casos excepcionales.
- Compare el número atómico ( Z ) de los átomos directamente unidos al estereocentro; el grupo que tiene el átomo de mayor número atómico recibe mayor prioridad.
- Si hay un empate, debemos considerar los átomos a una distancia 2 del estereocentro, ya que se hace una lista para cada grupo de átomos enlazados al que está directamente conectado al estereocentro. Cada lista está ordenada en orden decreciente de número atómico. Luego, las listas se comparan átomo por átomo; a la primera diferencia, el grupo que contiene el átomo de mayor número atómico recibe mayor prioridad.
- Si todavía hay un empate, cada átomo en cada una de las dos listas se reemplaza con una sublista de los otros átomos enlazados a él (a una distancia 3 del estereocentro), las sublistas se ordenan en orden decreciente de número atómico, y todo La estructura se compara de nuevo átomo por átomo. Este proceso se repite de forma recursiva, cada vez con átomos un enlace más lejos del estereocentro, hasta que se rompe el enlace.
Isótopos
Si dos grupos difieren solo en isótopos , entonces se usa la masa atómica más grande para establecer la prioridad.
Enlaces dobles y triples

Si un átomo A tiene un enlace doble con un átomo B, A se trata como si estuviera enlazado de forma simple a dos átomos: B y un "átomo fantasma" que es un duplicado de B (tiene el mismo número atómico) pero no está unido a nada. excepto A. Cuando B se reemplaza con una lista de átomos adjuntos, A en sí mismo, pero no su "fantasma", se excluye de acuerdo con el principio general de no duplicar un enlace que acaba de seguirse. Un triple enlace se maneja de la misma manera, excepto que A y B están conectados a dos átomos fantasmas del otro.
Isómeros geométricos
Si dos sustituyentes en un átomo son isómeros geométricos entre sí, el isómero Z tiene mayor prioridad que el isómero E.
Moléculas cíclicas
Para manejar una molécula que contiene uno o más ciclos , primero se debe expandir en un árbol (llamado dígrafo jerárquico ) atravesando enlaces en todos los caminos posibles comenzando en el estereocentro. Cuando el recorrido encuentra un átomo a través del cual ya ha pasado la ruta actual, se genera un átomo fantasma para mantener el árbol finito. Un solo átomo de la molécula original puede aparecer en muchos lugares (algunos como fantasmas, otros no) en el árbol.
Asignar descriptores
Estereocentros: R / S

Una vez que se han asignado sus prioridades a los sustituyentes de un estereocentro , la molécula se orienta en el espacio de modo que el grupo con la prioridad más baja se aleje del observador. Si los sustituyentes están numerados de 1 (prioridad más alta) a 4 (prioridad más baja), entonces el sentido de rotación de una curva que pasa por 1, 2 y 3 distingue los estereoisómeros . Un centro con sentido de rotación en el sentido de las agujas del reloj es un centro R ( recto ) y un centro con sentido de rotación en sentido antihorario es un centro S ( siniestro ). Los nombres se derivan del latín para 'derecha' e 'izquierda', respectivamente.
-1,2,3-trichlorocyclopentane.svg)
Un método práctico para determinar si un enantiómero es R o S es usando la regla de la mano derecha : uno envuelve la molécula con los dedos en la dirección 1 → 2 → 3 . Si el pulgar apunta en la dirección del cuarto sustituyente, el enantiómero es R ; de lo contrario, es S .
En casos raros, es posible que dos sustituyentes en un átomo difieran solo en su configuración absoluta ( R o S ). Si las prioridades relativas de estos sustituyentes necesitan ser establecidos, R tiene prioridad sobre S . Cuando esto sucede, el descriptor del estereocentro es una letra minúscula ( r o s ) en lugar de la letra mayúscula que se usa normalmente.
Enlaces dobles: E / Z
Para los alquenos y moléculas de doble enlace similares, se sigue el mismo proceso de priorización para los sustituyentes. En este caso, lo que importa es la colocación de los dos sustituyentes de mayor prioridad con respecto al doble enlace. Si ambos sustituyentes de alta prioridad están en el mismo lado del doble enlace, es decir, en la configuración cis , entonces al estereoisómero se le asigna una Z ( zusammen ). Si por el contrario están en una configuración trans , entonces al estereoisómero se le asigna una E ( entgegen ). En este caso, las letras de identificación se derivan del alemán para "juntos" y "opuesto", respectivamente.
Ejemplos de
Los siguientes son ejemplos de aplicación de la nomenclatura.
Asignaciones de R / S para varios compuestos 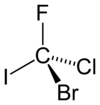
La molécula hipotética de bromoclorofluoroyodometano que se muestra en su configuración ( R ) sería un compuesto quiral muy simple. Las prioridades se asignan en función del número atómico ( Z ): yodo ( Z = 53)> bromo ( Z = 35)> cloro ( Z = 17)> flúor ( Z = 9). Permitir que el flúor (prioridad más baja) apunte lejos del espectador, la rotación es en el sentido de las agujas del reloj, por lo tanto, la asignación de R. 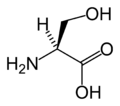
En la asignación de L -serina se le da la máxima prioridad al átomo de nitrógeno ( Z = 7) en el grupo amino (NH 2 ). Tanto el grupo hidroximetilo (CH 2 OH) como el grupo ácido carboxílico (COOH) tienen átomos de carbono ( Z = 6) pero se da prioridad a este último porque el átomo de carbono en el grupo COOH está conectado a un segundo oxígeno ( Z = 8 ) mientras que en el grupo CH 2 OH el carbono está conectado a un átomo de hidrógeno ( Z = 1). La prioridad más baja se da al átomo de hidrógeno y como este átomo de puntos lejos del espectador la disminución en sentido antihorario en prioridad sobre los tres sustituyentes restantes completa la asignación como S . 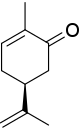
El estereocentro en ( S ) -carvona está conectado a un átomo de hidrógeno (no mostrado, prioridad 4) y tres átomos de carbono. El grupo isopropenilo tiene prioridad 1 (solo átomos de carbono) y para los dos átomos de carbono restantes la prioridad se decide con los átomos de carbono dos enlaces eliminados del estereocentro, una parte del grupo ceto (O, O, C, prioridad 2) y uno parte de un alqueno (C, C, H, prioridad 3). Los resultados rotación antihoraria resultantes en S .


-Carvone.svg)
Describiendo múltiples centros
Si un compuesto tiene más de un estereocentro quiral, cada centro se denota por cualquiera de R o S . Por ejemplo, la efedrina existe en los estereoisómeros (1 R , 2 S ) y (1 S , 2 R ), que son formas distintas de imagen especular entre sí, lo que los convierte en enantiómeros . Este compuesto también existe como los dos enantiómeros escritos (1 R , 2 R ) y (1 S , 2 S ), que se denominan pseudoefedrina en lugar de efedrina. Los cuatro de estos isómeros se denominan 2-metilamino-1-fenil-1-propanol en la nomenclatura sistemática. Sin embargo, la efedrina y la pseudoefedrina son diastereómeros o estereoisómeros que no son enantiómeros porque no están relacionados como copias en imagen especular. La pseudoefedrina y la efedrina reciben nombres diferentes porque, como diastereómeros, tienen propiedades químicas diferentes, incluso para mezclas racémicas de cada una.
Más generalmente, para cualquier par de enantiómeros, todos los descriptores son opuestos: ( R , R ) y ( S , S ) son enantiómeros, al igual que ( R , S ) y ( S , R ). Los diastereómeros tienen al menos un descriptor en común; por ejemplo ( R , S ) y ( R , R ) son diasteriómeros, al igual que ( S , R ) y ( S , S ). Esto también es válido para compuestos que tienen más de dos estereocentros: si dos estereoisómeros tienen al menos un descriptor en común, son diastereómeros. Si todos los descriptores son opuestos, son enantiómeros.
Cuando la asignación numérica de estereocentros no es única debido a la simetría especular de toda la molécula, el resultado es un mesocompuesto como el ácido mesotartárico , en el que ( R , S ) es lo mismo que la forma ( S , R ). En los compuestos meso, los estereocentros R y S ocurren en pares posicionados simétricamente.
Configuración relativa
La configuración relativa de dos estereoisómeros puede indicarse mediante los descriptores R y S con un asterisco (*). ( R *, R *) significa dos centros que tienen configuraciones idénticas, ( R , R ) o ( S , S ); ( R *, S *) significa dos centros que tienen configuraciones opuestas, ( R , S ) o ( S , R ). Para empezar, el centro estereogénico con el número más bajo (según la numeración sistemática de la IUPAC) recibe el descriptor R *.
Para designar dos anómeros se utilizan los estereodescriptores relativos alfa (α) y beta (β). En el anómero α, el átomo de carbono anomérico y el átomo de referencia tienen configuraciones opuestas ( R , S ) o ( S , R ), mientras que en el anómero β son iguales ( R , R ) o ( S , S ).
Caras

La estereoquímica también juega un papel en la asignación de caras a moléculas trigonales como las cetonas . Un nucleófilo en una adición nucleófila puede acercarse al grupo carbonilo desde dos lados o caras opuestos. Cuando un nucleófilo aquiral ataca a la acetona , ambas caras son idénticas y solo hay un producto de reacción. Cuando el nucleófilo ataca a la butanona , las caras no son idénticas ( enantiotópicas ) y resulta un producto racémico . Cuando el nucleófilo es una molécula quiral , se forman diastereoisómeros . Cuando una cara de una molécula está protegida por sustituyentes o restricciones geométricas en comparación con la otra cara, las caras se denominan diastereotópicas . Las mismas reglas que determinan la estereoquímica de un estereocentro ( R o S ) también se aplican al asignar la cara de un grupo molecular. Las caras se denominan Re -face y Si -face . En el ejemplo que se muestra a la derecha, el compuesto acetofenona se ve desde el Re -face. La adición de hidruro como en un proceso de reducción de este lado formará el enantiómero ( S ) y el ataque de la cara de Si opuesta dará el enantiómero ( R ). Sin embargo, se debe tener en cuenta que agregar un grupo químico al centro proquiral desde el Re -face no siempre conducirá a un estereocentro ( S ), ya que debe tenerse en cuenta la prioridad del grupo químico. Es decir, la estereoquímica absoluta del producto se determina por sí sola y no considerando desde qué cara fue atacado. En el ejemplo mencionado anteriormente, si se añadiera cloruro ( Z = 17) al centro proquiral del Re -face, esto daría como resultado un enantiómero ( R ).